My research focuses on the development of models and efficient algorithms for mining, integrating and comparing systems-level, large, complex and heterogeneous networked data. I am applying these methods on molecular and clinical data to yield new insight into problems related to human health, and on world trade data for tracking the dynamics of economic systems.
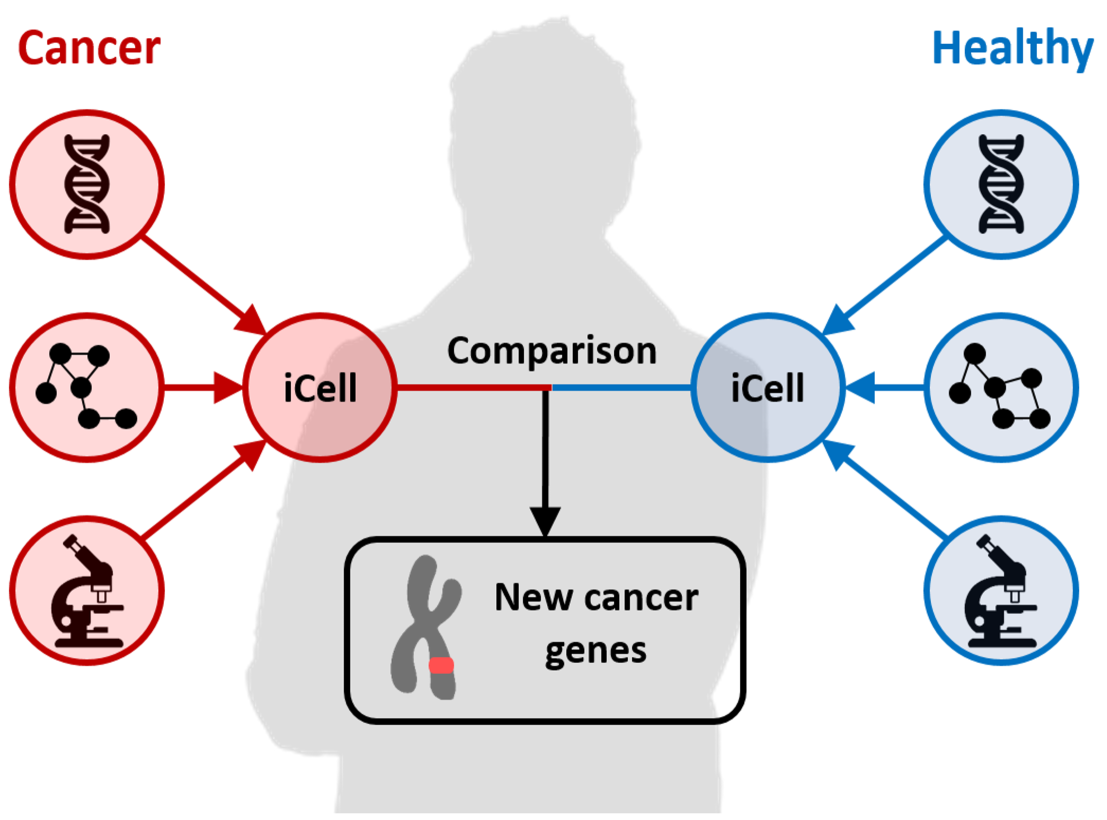
Figure: the iCell framework. We proposed the integrated cell (iCell) methodology to integrate and compare tissue-specific molecular networks [J20]. We first applied it on cancer datasets, uncovering new cancer-genes that were biologically validated (wet-lab). We extended this model to uncover Covid-19 related genes [J34] and to study the scenescence escape mechanisms of drug resistant melanoma [J37].
Research axes
We are flooded with large scale omics data that each represents a partial view on the molecular functioning of the cell. In order to gain novel knowledge, we develop data-integration frameworks to mine these datasets collectively rather than in isolation from each other's, yielding new insight into diseases by simultaneously stratifying patients into clinically relevant sub-groups, uncovering new disease-related genes, and predicting drug-repurposing. While we mainly focus on cancer (e.g., [J20, J37]), we also apply our methodologies to study Covid-19 [J30, J34], rare diseases [J36] and Parkinson's disease.
Simple models such as networks, which can only represent pairwise relationships, cannot fully capture the higher organization of biological processes in the cell.
To comprehensively capture the organization of complex biological systems, we propose to model those using simplicial complexes [J19] from computational geometry or by using hypernetworks [J18] from graph theory, and we extend our network analytics tools to mine the information hidden in these new models.
In the cell, molecules (e.g., proteins) do not act in isolation but rather interact with other molecules in order to perform their functions. The same holds in other fields that study interacting objects, such as in economics, where countries trade with each other's. To uncover domain specific knowledge, we develop tools to characterized the wiring patterns of nodes in networks, yielding new insight on the core-broker-periphery organization of the world trade network [J6, J13] and in the topological conservation of biological functions in protein-protein interaction networks [J6, J7, J13].
We also develop pairwise and multiple global network alignment methods, which allow us to uncover evolutionary relationships between species and to transfer biological annotations across the networks of different species [J8, J9, J15].
There is an increasing gap between captured proteins and available annotations. because structurally similar proteins tend to share biological functions, we propose methods for comparing the 3D structures of isolated proteins [J1, J2, J3, J5], which allows for transferring functional annotations across proteins.
Moreover, since protein does not act in isolation but bind with other molecules (ligands), we develop topological and geometrical descriptors of protein binding interfaces that allow predicting the protein-ligand binding affinity [J4]. This is of first importance when developing new drugs, which should have higher affinities with their target proteins than the natural ligands.