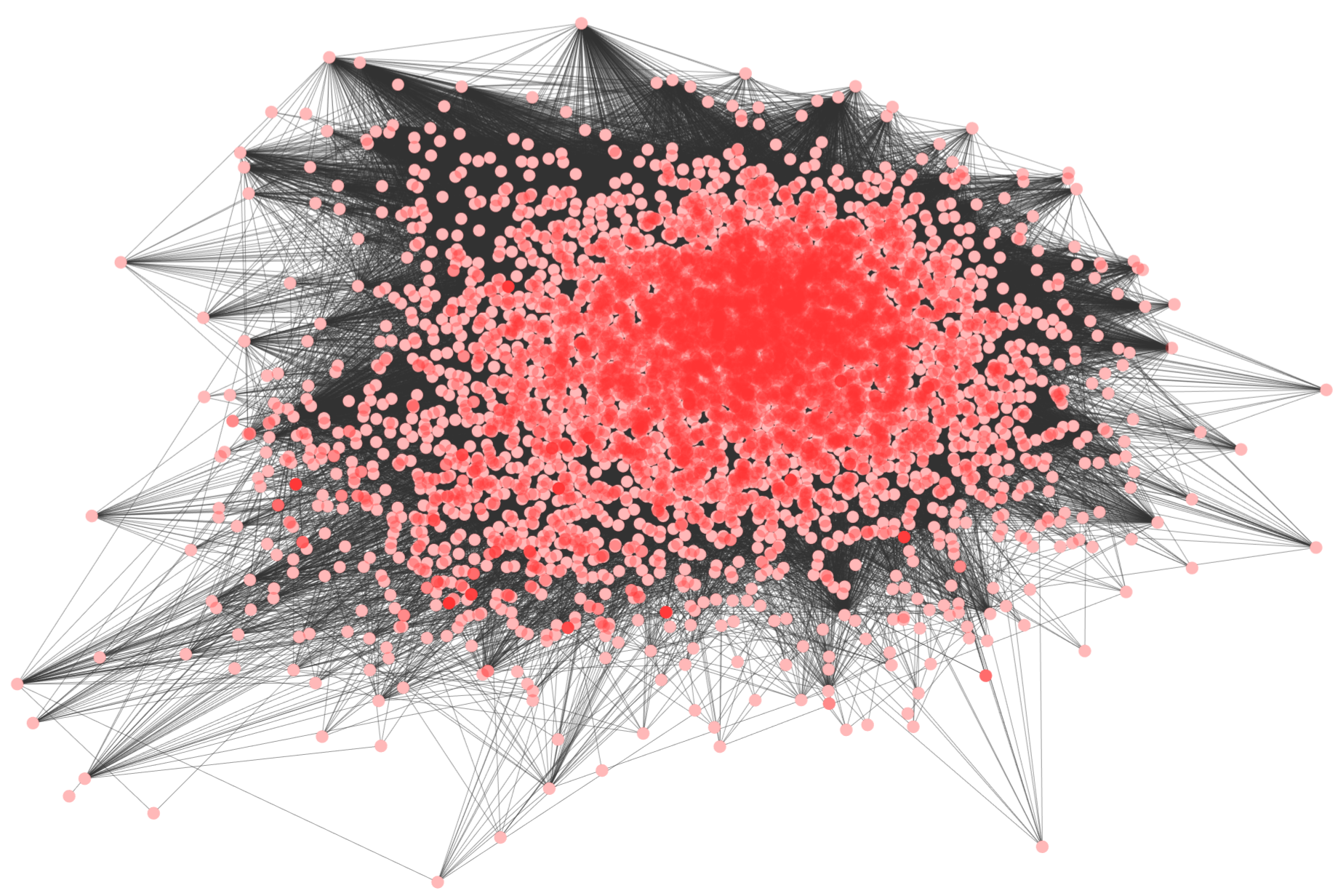
Alignment-free network comparison; We use graphlets (small induced sub-graphs) and their statistics to measure the similarities between networks. Our tools are available for DIRECTED networks and for UNDIRECTED networks.
FUSE; FUSE is a novel global multiple network aligner. It combines novel similarity scores between proteins, which are obtained by fusing all PPI networks being aligned using non-negative matrix tri-factorization (NMTF), with a novel approximate maximum weight k-partite matching solver. FUSE is available here.
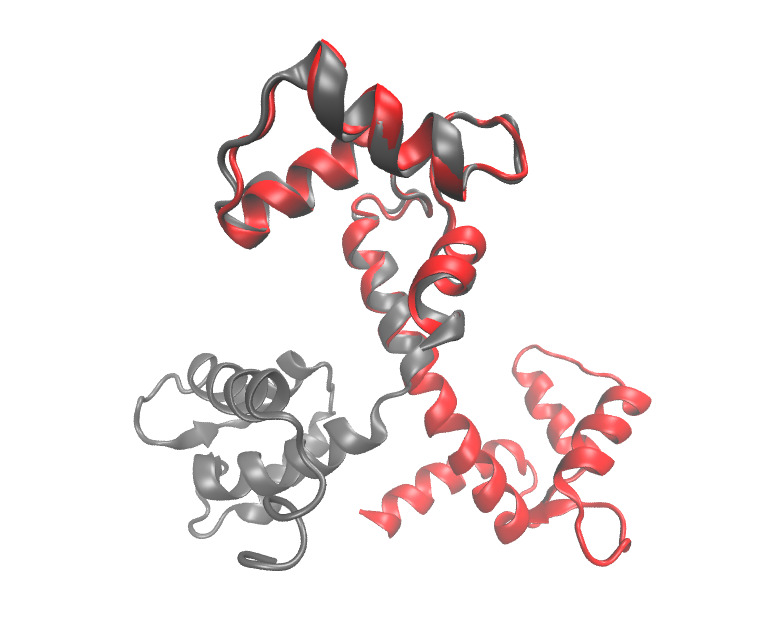
L-GRAAL; L-GRAAL is a new pairwise network aligner. L-GRAAL uses a novel scoring functions that combines the homological relationships of the mapped proteins (i.e., their sequence similarities) with the topological similarities of the mapped interactions (based on the graphlet degree similarity). L-GRAAL search for an alignment that maximise this scoring function using a novel search algorithm based on Lagrangian Relaxation. L-GRAAL is available here.
Addict; Tools for solving the interval Stoichiometry Determination (SD) problem, computing the Frobenius number associated to the input masses, and estimating upper and lower bounds on the number of solutions of a given interval SD problem. Addict is now part of the Structural Bioinformatics Library (SBL).
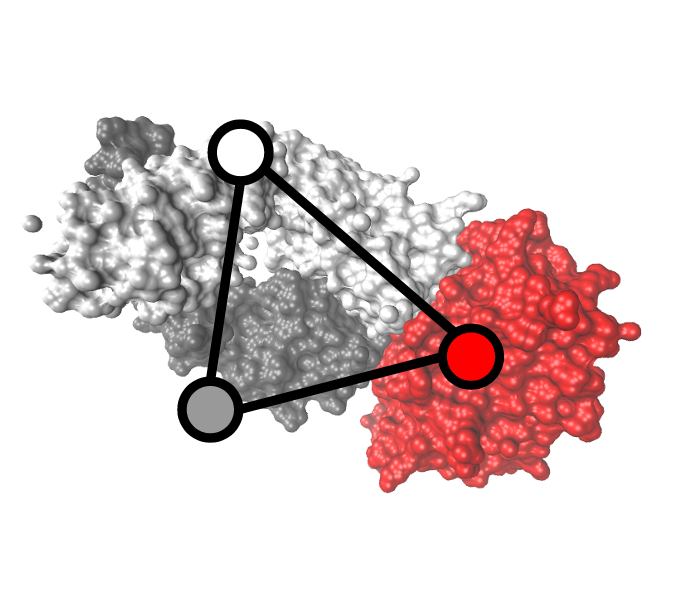
Vorpatch and Compatch; Vorpatch computes the two Atom Shelling Tree (AST) models of a protein complex interface. Then, Compatch allows comparing ASTs using a generic tree-edit-distance algorithm, which is instantiated to favour topological or geometrical similarity. Both software are now parts of the Structural Bioinformatics Library (SBL).
GR-Align; Maximum Contact Map Overlap (CMO) is a flexible protein structure comparison paradigm that received sustained attention during the last decades. GR-Align is a CMO heuristic based on graphlets (small induced sub-graphs) that is suited for database searches. GR-Align is available here.
CSA; Comparative Structural Alignment is a web server for the computation, evaluation and comprehensive comparison of pairwise protein structure alignments. Access CSA web-server.
A_purva; A flexible protein structure comparison tool, based on the maximum contact map overlap approach. A_purva can be downloaded as a part of the Structural Bioinformatics Library (SBL).