Ph.D in Computer Science at the University of Rennes 1, with high Honors
Title: "Protein Structure Comparison: From Contact Map Overlap Maximisation to Distance-based Alignment Search Tool"
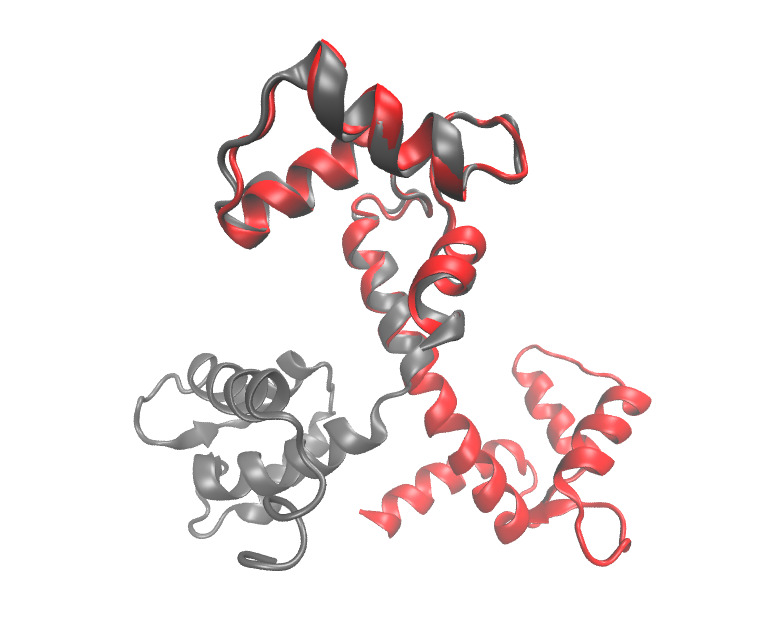
Abstract: In structural biology, it is commonly admitted that the three dimensional structure of a protein determines its function. A fruitful assumption based on this paradigm is that proteins sharing close three dimensional structures may derive from the same ancestor and thus, may share similar functions. Computing the similarity between two protein structures is therefore a crucial task and has been extensively investigated. Among all the proposed methods, we focus on the similarity measure called Contact Map Overlap maximisation (CMO), mainly because it provides scores which can be used for obtaining good automatic classifications of the protein structures.
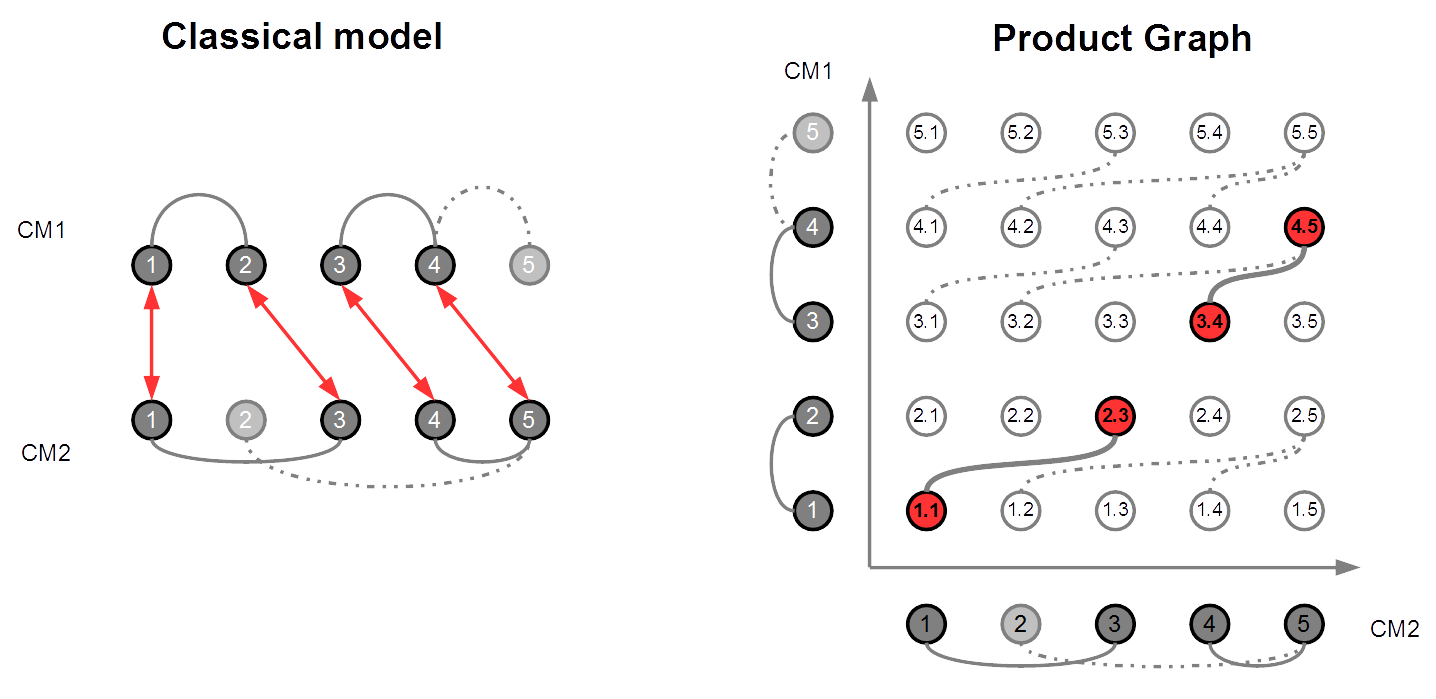
In this thesis, comparing two protein structures is modelled as finding specific sub-graphs in specific k-partite graphs called alignment graphs, and we show that this task can be efficiently done by using advanced combinatorial optimisation techniques. In the first part of the thesis, we model CMO as a kind of maximum edge induced sub-graph problem in alignment graphs, for which we conceive an exact solver which outperforms the other CMO algorithms from the literature. Even though we succeeded to accelerate CMO, the procedure still stays too much time consuming for large database comparisons. The second part of the thesis is dedicated to further accelerate CMO by using structural biology knowledge. We propose a hierarchical approach for CMO which is based on the secondary structure of the proteins. Finally, although CMO is a very good scoring scheme, the alignments it provides frequently posses big root mean square deviation values. To overcome this weakness, in the last part of the thesis, we propose a new comparison method based on internal distances which we call DAST (for Distance-based Alignment Search Tool). It is modelled as a maximum clique problem in alignment graphs, for which we design a dedicated solver with very good performances.
Keywords: protein structure comparison, contact map overlap maximisation, k-partite graph, maximum edge induced sub-graph, maximum clique, integer programming, branch and bound.